理論
Advance/OF-DFTに実装されているAtomic Orbital Facilitated DFT (AOF-DFT)と、それに関連する手法について、理論を解説します。
Orbital-Free DFT (OF-DFT)
密度汎関数理論(Density Functional Theory, DFT)は、系の電子密度  の汎関数としてエネルギー
の汎関数としてエネルギー ![E[\rho]](_images/math/22edfaf664b837608340b2944e34723b7c6e5b1f.svg) が与えられ、エネルギーを最小化することで基底状態の電子密度が得られるというHohenberg-Kohnの定理に基づきます [1]。
が与えられ、エネルギーを最小化することで基底状態の電子密度が得られるというHohenberg-Kohnの定理に基づきます [1]。
DFTに基づく第一原理電子状態計算の標準的な手法となっているKohn-Sham DFT (KS-DFT)では、仮想的な参照系として相互作用のないKohn-Sham系を導入し、エネルギー汎関数の運動エネルギー(Kinetic Energy)項の計算にKS系の波動関数(Kohn-Sham軌道)  を使います [2]。
を使います [2]。
(1)![& E[\rho] = \underset{\mathrm{Kinetic}}{-\frac{1}{2}\sum_{i}\langle\psi_i\lvert\nabla^2\rvert\psi_i\rangle} + \underset{\mathrm{External\ Potential}}{E_\mathrm{ext}[\rho]} + \underset{\mathrm{Hartree}}{E_\mathrm{H}[\rho]} + \underset{\mathrm{Exchange\ Correlation}}{E_\mathrm{XC}[\rho]}](_images/math/24fe1ca822c418acdaaa5c1faa460c6f8fc45ecc.svg)
KS軌道はKohn-Sham方程式と呼ばれる方程式の固有関数であり、KS-DFTでは固有値問題を数値的に解く必要があります。これが原子数  に対し計算量が
に対し計算量が  となり、「第一原理計算が重い」と言われる理由です。
となり、「第一原理計算が重い」と言われる理由です。
対して、運動エネルギーの計算にKS軌道を使わず(Orbital-Free)、電子密度の汎関数である運動エネルギー汎関数 ![T[\rho]](_images/math/43b160d3b6c5e240cd7c9b3601f1e6b58c1924c2.svg) を用いて計算量を
を用いて計算量を  に抑えよう、というのがOrbital-Free DFT (OF-DFT)の考え方です。
に抑えよう、というのがOrbital-Free DFT (OF-DFT)の考え方です。
この考え方は実用上、とても重要なものです。例えば、対象とする系の原子数 が10倍になった場合を考えると、KS-DFTでは計算時間が約1000倍に跳ね上がるのに対し、OF-DFTであれば10倍程度の増加に抑えられます。この差は原子数が大きくなるほど顕著になり、KS-DFTでは実質的に不可能な大規模システムのシミュレーションもOF-DFTでは可能となります。
以下に、KS-DFTとOF-DFTに加えて、近年注目されているグラフニューラルネットワーク(GNN)力場などの機械学習力場を用いる力場法を比較します。
特徴 \ 手法 |
KS-DFT |
OF-DFT |
力場法 |
|---|---|---|---|
電子密度 |
あり |
あり |
なし |
軌道 |
あり |
なし |
なし |
計算精度 |
高い |
汎関数に依存 |
力場に依存 |
計算コスト |
|
|
|
汎用性 |
全元素に適用可能 |
擬ポテンシャルによる制約 |
力場に依存 |
力場法と比較すると、OF-DFTは計算コストが低水準の で同じでありながら、電子密度を明示的に扱うことにより、外部電場の印加や電子・ホールのドープ、仕事関数等の計算ができるという大きな特長があります。
OF-DFTは、以上のような長所をもつ一方で、その精度を左右する運動エネルギー汎関数の厳密な式は知られていません。近似や機械学習 [3] [4] による運動エネルギー汎関数の研究が進められるも、実用に耐えうる計算精度・汎用性は実現されませんでした。
さらに、OF-DFTには、適用可能な擬ポテンシャルが充実していないという問題もあります。OF-DFTでは、波動関数に依存した非局所演算子が使用できないため、KS-DFT用のノルム保存擬ポテンシャルやウルトラソフト擬ポテンシャルは利用できません。局所項のみで構成された局所擬ポテンシャルを使う必要があり、例えば代表的なものの一つであるOptimized Effective Pseudo Potential [5] では、27種類の元素にしか対応することができません。
そのような中で、弊社独自開発の汎グラフを応用した深層学習運動エネルギー汎関数AdvanceSoft26は、その高い精度により、実用に耐えうるOF-DFT計算を実現しました。 さらに、独自にOF-DFTを拡張したAOF-DFTという手法により、一般に広く使われるKS-DFT用ノルム保存擬ポテンシャルの適用も可能としました。
汎グラフによる深層学習運動エネルギー汎関数
OF-DFTの機械学習運動エネルギー汎関数の開発において、連続的な場である電子密度に対し機械学習を行うには、特徴ベクトルをどのように抽出するかが問題になります。既報の研究では、座標  における特徴ベクトルをあらかじめ定義しておくという方法を使っていますが、十分な精度や汎用性が担保できませんでした。
における特徴ベクトルをあらかじめ定義しておくという方法を使っていますが、十分な精度や汎用性が担保できませんでした。
一方、機械学習力場の分野では、グラフ理論を応用して、隣接原子間の相互作用を模した学習可能な特徴ベクトルを使用するGNN力場が上手くいっています。グラフは離散的なノード・エッジから構成されますが、このような学習可能な特徴ベクトルを機械学習汎関数にも応用できないか、という考えから、弊社ではグラフを連続多変数空間に拡張したものを独自に開発し、これを汎グラフと名付けました。関数(Function)→汎関数(Functional)に倣い、グラフ(Graph)→汎グラフ(Graphical)としています。
まず、汎グラフの着想元となる、通常のグラフ上のグラフ畳み込みネットワーク(Graph Convolutional Network, GCN)について説明します。ノード  と、それらを繋ぐエッジを考えます。各エッジにはノード間の繋がりを表す係数
と、それらを繋ぐエッジを考えます。各エッジにはノード間の繋がりを表す係数  を設定します。各ノードに初期値(入力)
を設定します。各ノードに初期値(入力)  を与え、この値(特徴量)を更新していくことを考えます。
を与え、この値(特徴量)を更新していくことを考えます。
更新は、「隣接ノードの特徴量の集約」と「重みによる変換」の2段階で行います。
(2)
(3)
 は非線形な振る舞いを表現するために使われる活性化関数です。また、
は非線形な振る舞いを表現するために使われる活性化関数です。また、  を
を  に対する差分の形で更新する方法を残差接続と呼びます。
に対する差分の形で更新する方法を残差接続と呼びます。
このままでは更新を繰り返しても特徴量を拡散させるフィルタのようにしか働きません。そこで、マルチチャンネル化を行います。特徴量をチャンネル数の次元を持つ特徴ベクトル  とし、更新には重み行列
とし、更新には重み行列  を使います。
を使います。
(4)
特徴ベクトルの各チャンネルの混合を含めた処理が入り、この更新(畳み込み)を繰り返すことにより、複雑な処理を行えるようになります。最終的に目的の特徴ベクトル(出力)が得られるように  を最適化するのが、GCNの学習ということになります。
を最適化するのが、GCNの学習ということになります。
次に、GCNを連続多変数空間上で定義された場に適用できるよう拡張することを考えます。このとき、グラフと汎グラフの対応関係は以下の表のようになります。
グラフ |
汎グラフ |
|---|---|
ノード |
座標 |
エッジ |
座標の組 |
特徴ベクトル |
場 |
隣接行列 |
隣接相関 |
特徴量の集約 |
隣接相関を積分核とする畳み込み積分 |



隣接相関  は定義上は
は定義上は  で非ゼロの値を持つデルタ関数になりますが、数値計算を行うために、動径方向に幅を持つガウス関数で置き換えます。角度方向には球対称とし、球平均を取って距離のみの関数にします。
で非ゼロの値を持つデルタ関数になりますが、数値計算を行うために、動径方向に幅を持つガウス関数で置き換えます。角度方向には球対称とし、球平均を取って距離のみの関数にします。
(5)
半径  、幅
、幅  がパラメータとなります。種々の
がパラメータとなります。種々の  対に対する
対に対する  を使って特徴量を集約することで、空間全体をカバーするようなマルチチャンネル化を行ったことになります。また、畳み込み積分の距離を有限にするため、カットオフ関数
を使って特徴量を集約することで、空間全体をカバーするようなマルチチャンネル化を行ったことになります。また、畳み込み積分の距離を有限にするため、カットオフ関数  を適用します。
を適用します。
最終的に、汎グラフは次のように定式化されます。
(6)
(7)
が連続のままであることに注目してください。数値計算を行う上では離散的な (メッシュ)を使いますが、汎グラフのパラメータはメッシュの細かさに依存しません。学習時と推論時でメッシュの細かさや形が異なっていても良く、transferabilityが高いという特長があります。
この汎グラフを適用した深層学習運動エネルギー汎関数AdvanceSoft26 (AS26)の基本的なアーキテクチャは下図のようになります。汎グラフに入力する特徴ベクトルは、電子密度の勾配とラプラシアンからつくられます。
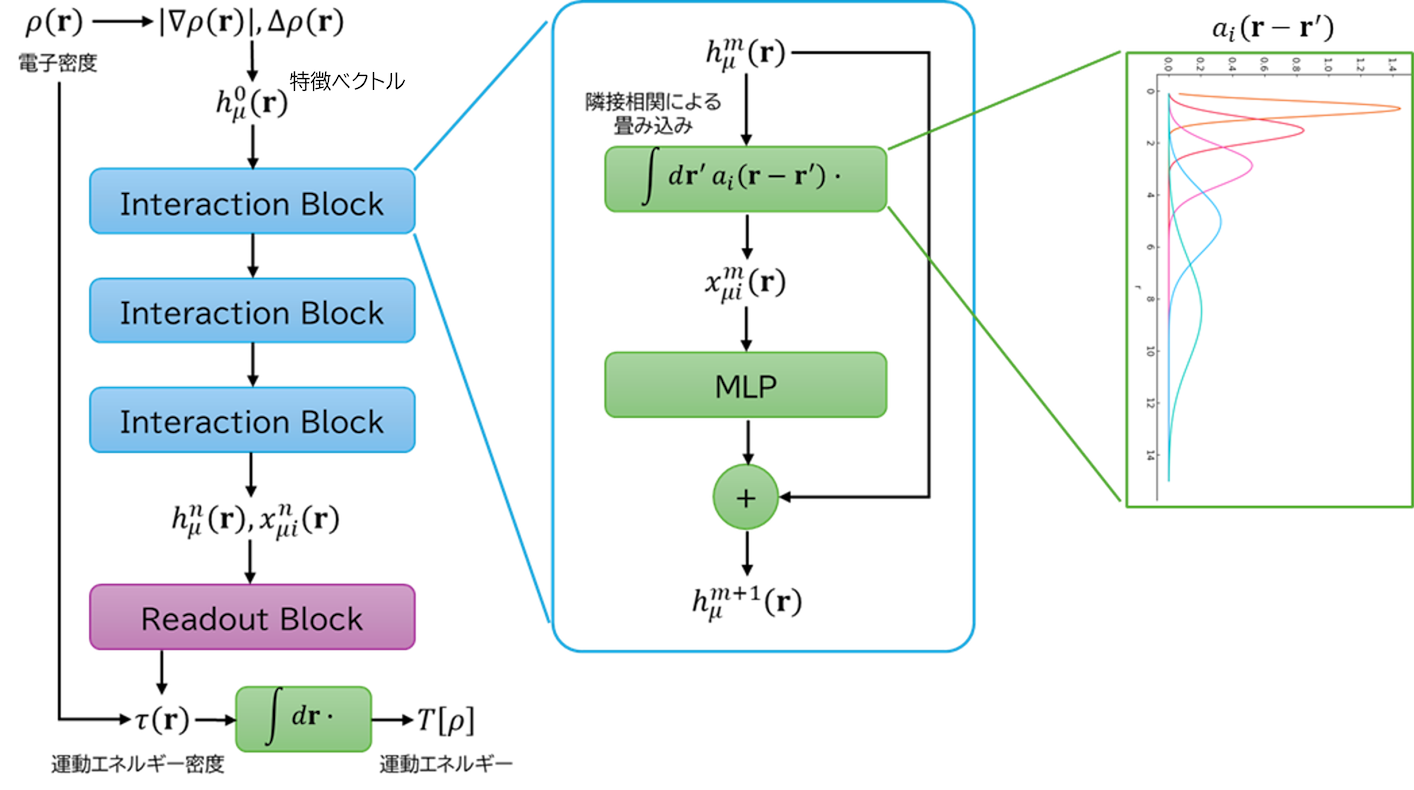
出力は、Thomas-Fermi (TF)モデルおよびvon Weizsäcker (vW)モデル [3] で計算した運動エネルギー密度  に対する補強因子
に対する補強因子 ](_images/math/c557d5ee458b33dda84cc8d2f618e5a6cb366d7a.svg) として使い、最終的な運動エネルギーを得ます。
として使い、最終的な運動エネルギーを得ます。
(8)![T_\mathrm{AS26}[\rho] = \int \mathrm{d}\mathbf{r} \lbrace\tau^\mathrm{TF}(\mathbf{r})F_\mathrm{TF}^\mathrm{NN}[\rho](\mathbf{r}) + \tau^\mathrm{vW}(\mathbf{r})F_\mathrm{vW}^\mathrm{NN}[\rho](\mathbf{r})\rbrace](_images/math/167bec3f9697f7313f6b3d4c25d91f11c3e1f717.svg)
さらに、分子系に対しては以下の非局所のWang-Teter(WT)モデル [3] の項を追加します。
(9)
ここで、  はフェルミ波数
はフェルミ波数  の総数であり、Wang-Teterの運動エネルギー密度
の総数であり、Wang-Teterの運動エネルギー密度  はGELU関数によって正負の成分に分離されています。
はGELU関数によって正負の成分に分離されています。
以上のようにして開発された汎グラフによる深層学習運動エネルギー汎関数は、OF-DFTにおいて、従来の運動エネルギー汎関数を大きく上回る精度と汎用性を実現しました。
局所密度行列汎関数理論 (L-DMFT)
先述の通り、OF-DFTは、波動関数に依存した非局所演算子が使用できないことからKS-DFT用のノルム保存擬ポテンシャルが利用できず、適用元素が限定されます。
弊社ではこの課題を解決すべく、原子ごとの局所的な密度行列は厳密に扱い、原子間は電子密度の汎関数で計算する局在密度行列汎関数理論 (Localized Density Matrix Functional Theory, L-DMFT)という手法を開発しました。
L-DMFTは、OF-DFTをDMFTにより拡張した理論であり、基底状態のエネルギー  を、電子密度 だけではなく、1電子縮約密度行列
を、電子密度 だけではなく、1電子縮約密度行列  の汎関数とします。
の汎関数とします。
(10)![E[\rho, \tilde{\rho}] = \underset{\mathrm{Kinetic}}{T[\rho]} + \int\underset{\mathrm{Local\ potential}}{ d\mathbf{r} \, v_{\mathrm{loc}}(\mathbf{r}) \rho(\mathbf{r})} + \underset{\mathrm{Non-local\ potential}}{E_{\mathrm{NL}}[\tilde{\rho}]} + \underset{\mathrm{Hartree,\ Exchange\ Correlation}}{E_\mathrm{HXC}[\rho]}](_images/math/cdc1a54b8fa8647dde42dec1cfe7d54336cf125c.svg)
 は局所ポテンシャル、
は局所ポテンシャル、 は非局所エネルギーです。密度行列 は非局所エネルギー の計算にのみ使われるため、原子核の座標
は非局所エネルギーです。密度行列 は非局所エネルギー の計算にのみ使われるため、原子核の座標  からみたカットオフ半径
からみたカットオフ半径  の内側のみに限定します。
の内側のみに限定します。
(11)
(12)
非局所エネルギー 以外の部分は、従来のOF-DFTと同様に電子密度 のみで計算可能です。
以上のようにして、各原子に局在化した密度行列を用いることによって、非局所演算子の計算が可能となり、KS-DFT用のノルム保存擬ポテンシャルの適用が実現します。 また、純粋なOF-DFTでは難しかったDFT+U(電子相関の補正)やスピン軌道相互作用といった局所的な物理現象の計算も実装可能となります。
さらに、従来のOF-DFTに比べて情報量および演算量は増えるものの、その量は原子数に比例する程度であり、計算コストは低水準の に維持することができます。
Atomic Orbital Facilitated DFT (AOF-DFT)
L-DMFTを、擬原子軌道を用いて解く手法をAtomic Orbital Facilitated DFT (AOF-DFT)と呼称します。
AOF-DFTでは、L-DMFTの定義を満たすように、1電子縮約密度行列 および電子密度 を以下のように定義します。
(13)
(14)
ここで、 は
は  番目の原子における
番目の原子における  番目の擬原子軌道、
番目の擬原子軌道、 は原子内の成分のみで構成されたブロック対角密度行列です。
は原子内の成分のみで構成されたブロック対角密度行列です。
式 (13) , (14) を式 (10) のエネルギー汎関数 に代入し、以下の式 (15) , (16) で表される 表示可能性の条件および電子数保存則のもとで密度行列 を変分原理にて最適化することで、基底状態を求めることができます。
(15)
(16)
ここで、 は軌道占有数、
は軌道占有数、 は擬原子軌道の展開係数であり、同一原子内で原子軌道 が規格直交化されていると仮定しました。
は擬原子軌道の展開係数であり、同一原子内で原子軌道 が規格直交化されていると仮定しました。
式 (13) , (14) , (15) を用いて、式 (10) のエネルギー汎関数 の変分をとると、次のような に対する「原子毎の」固有値問題に帰着します。
(17)
(18)![\begin{aligned}
H_{ij}^A = &\int d\mathbf{r} \chi_i^A (\mathbf{r}) \left[ \frac{\delta T}{\delta \rho(\mathbf{r})} + \frac{\delta E_{\mathrm{HXC}}}{\delta \rho(\mathbf{r})} + v_{\mathrm{loc}} (\mathbf{r}) \right] \chi_j^A (\mathbf{r}) \\
&+ \sum_B \sum_k \left[ \int d\mathbf{r} \chi_i^A (\mathbf{r}) \beta_k^B (\mathbf{r}) \right] D_k^B \left[ \int d\mathbf{r}' \chi_j^A (\mathbf{r}') \beta_k^B (\mathbf{r}') \right]
\end{aligned}](_images/math/32c822ff6a0e709d9297d5e43a0ece6fed865e33.svg)
ハミルトニアン  の右辺第2項が、非局所エネルギーに相当します。
の右辺第2項が、非局所エネルギーに相当します。 は
は  番目の原子の
番目の原子の  番目の射影演算子、
番目の射影演算子、 は各射影演算子に対するエネルギーです。
は各射影演算子に対するエネルギーです。 の場合も有限な寄与があるため計算に含めますが、これはカットオフ半径に重なりがある場合に限られるため、計算コストは低水準の に維持されます。
の場合も有限な寄与があるため計算に含めますが、これはカットオフ半径に重なりがある場合に限られるため、計算コストは低水準の に維持されます。
原子毎の固有値方程式 (式 (17))を解くと同時に、電子数保存則 (式 (16))を満たすように全ての原子の軌道占有数 を決定し、最終的に式 (15) の密度行列を得ます。
AOF-DFTでは、OF-DFTの場合と同様に、既存の運動エネルギー汎関数を用いると実用に耐えうる精度は担保されませんが、深層学習運動エネルギー汎関数を用いることでこの問題を解決できます。
深層学習運動エネルギー汎関数とAOF-DFTの開発により、OF-DFTの の計算コストを維持したまま、実用に耐えうる精度で、全元素対応の電子状態計算が可能となりました。
運動エネルギーの空間分割
原子核近傍においては、価電子軌道であっても比較的に大きな振幅を持つため、運動エネルギーは非常に大きくなります。このように大きな振幅を持つ軌道に対しては、運動エネルギーを汎関数として表現するのは容易ではありません。そこでAOF-DFTでは、原子核近傍に適当な重み関数を設定しておき、重み関数の存在する部分についてのみ、汎関数だけではなく運動エネルギー演算子も用いて厳密に計算するというアプローチをとっています。
具体的には、運動エネルギー  を、原子核近傍の運動エネルギー
を、原子核近傍の運動エネルギー  と、汎関数による運動エネルギー
と、汎関数による運動エネルギー  の和とします。
の和とします。
(19)
原子核近傍の運動エネルギー は以下のように定義します。
(20)
ここで、  は原子 に局在化した重み関数であり、原子核近傍で1、遠方で0の値をとります。
式 (13) を用いて式 (20) 展開すると、次のようになります。
は原子 に局在化した重み関数であり、原子核近傍で1、遠方で0の値をとります。
式 (13) を用いて式 (20) 展開すると、次のようになります。
(21)
一方、運動エネルギー汎関数の寄与 は、重み関数 と運動エネルギー密度 を用いて、以下のように定義します。
(22)
例えば、運動エネルギー汎関数としてAS26を用いる場合、 は以下のようになります。
(23) + \tau^\mathrm{vW}(\mathbf{r})F_\mathrm{vW}^\mathrm{NN}[\rho](\mathbf{r})](_images/math/26365631193b6b87c6094130975cb9a20b03acde.svg)
現行のバージョンのAOF-DFTでは、以下で定義される重み関数  を使用しています。
を使用しています。
(24)
ここで、  は原子 の中心からの距離であり、
は原子 の中心からの距離であり、  の値としては、原子 の共有結合半径の1.2倍の値を用いています。
の値としては、原子 の共有結合半径の1.2倍の値を用いています。
Preconditioning演算子
一般に、機械学習運動エネルギー汎関数の汎関数微分  は、高周波領域にて不安定である(ノイズを含む)ことが知られています [6] 。汎関数微分 は電子密度を更新する際に用いるポテンシャルに相当するため、機械学習運動エネルギー汎関数を用いるOF-DFTのSCF計算は不安定となります。
は、高周波領域にて不安定である(ノイズを含む)ことが知られています [6] 。汎関数微分 は電子密度を更新する際に用いるポテンシャルに相当するため、機械学習運動エネルギー汎関数を用いるOF-DFTのSCF計算は不安定となります。
AOF-DFTにおいても同様に存在するこの問題を解決すべく、KS-DFTにおける電荷密度混合法や波動関数対角化にも使用されるPreconditioning演算子  を導入しました。
を導入しました。
(25)
ただし、AOF-DFTにおけるPreconditioning演算子は高周波成分を除去する必要があるため、KS-DFTにおけるKerker演算子などとは異なる定義式が要求されます。そこで、具体的には、以下の5つの定義を用意しました。
- Gaussian Preconditioner
![K(\mathbf{g}) = \exp \left[ -\frac{g^2 \sigma^2}{2} \right]](_images/math/b3e1e41d505f507f47d2b0ed733bb63b678c01dc.svg)
標準偏差
のガウス関数による、位置 における  の球平均に相当します。
の球平均に相当します。- Lorentz Preconditioner

Gaussian Preconditionerの指数関数版に相当します。
 は波数ベクトルのダンピング因子です。
は波数ベクトルのダンピング因子です。- Teter Preconditioner

VASPのRMM-DIISなどで波動関数に対して利用される演算子です [7]。
- Damping Preconditioner

TD-DFTやRISMで使用されるダンピング関数です [8] 。
- Two-Thirds Preconditioner

電子密度のカットオフエネルギー
 に基づく最大波数の2/3をカットオフ波数
に基づく最大波数の2/3をカットオフ波数  とする演算子です [9] 。
とする演算子です [9] 。